
Webpage Menu:
⚃ 2.2.4.1 Introduction:
Click for updated 2024 information on IBM classification.⚃ 2.2.4.2 Myositis-specific auto-antibodies (MSAs):
⚃ 2.2.4.1 Introduction:
⚄ 2.2.4.1.1 Muscle diseases have traditionally been put
under the umbrella of
myopathy:
a disease of the muscle that causes weakness of the muscle (
not a disease involving the nerves
).
≻ As discoveries are made, the classification evolves, but there is
no currently accepted international classification system for muscle
diseases.
≻ As a group, these diseases are sometimes referred to as muscular
dystrophy diseases.
≻ Inclusion body myositis is usually listed under the umbrella of
muscular dystrophy diseases.
≻ A second Umbrella is the idiopathic inflammatory myopathies (IIM):
a group of conditions characterised by inflammation of muscles (myositis)
and other body systems.
≻≻ This is where IBM has traditionally been classified along
with polymyositis (PM) and dermatomyositis (DM), but the features of IBM
are
clearly distinct
from these other two types.
Figure 2.2.4.1.1a
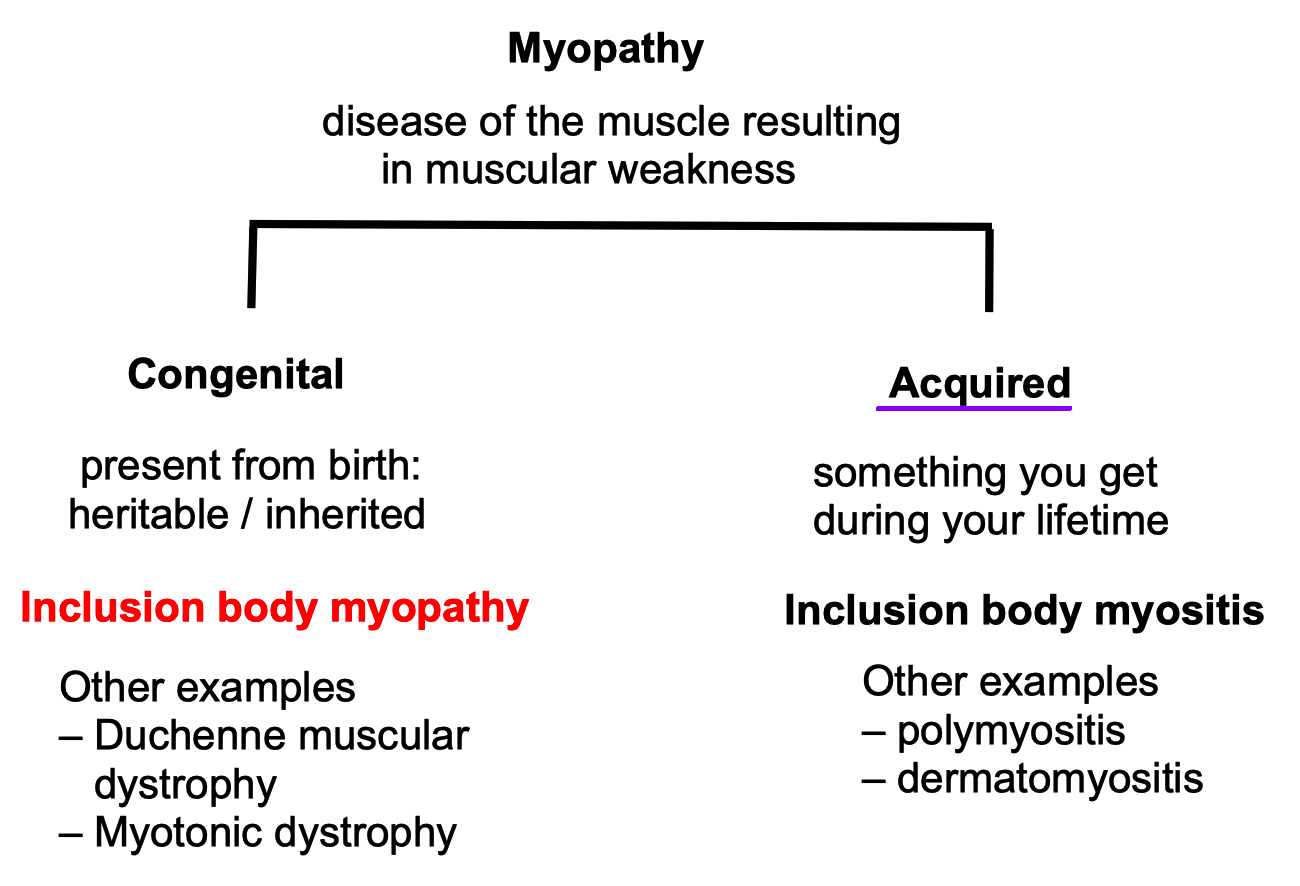
Figure 2.2.4.1.1b
⚄ 2.2.4.1.2 Myositis versus myopathy.
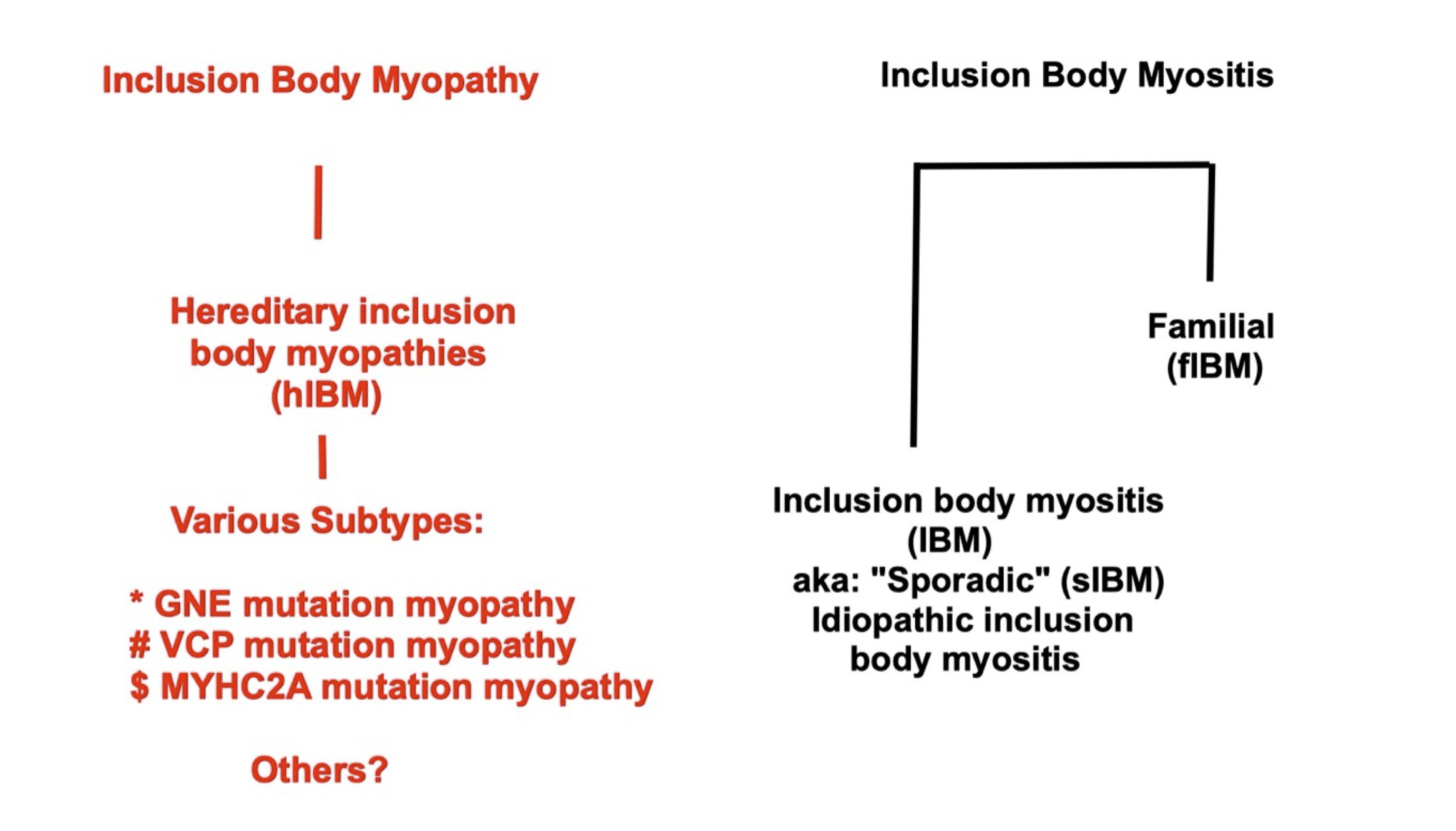
Figure 2.2.4.1.2
⚃ Notes on Figure 2.2.4.1.2
⚄ IBM is an abbreviation for 'inclusion body myositis' not
'inclusion body myopathy' (Greenberg, 2019, Milone, 2017).
≻ The abbreviation IBM
should
refer clearly and only to the single disease inclusion body myositis, not
to the hereditary inclusion body myopathies (“hIBMs”).
≻ On this page, IBM will only refer to inclusion body myositis and I
will not specify sporadic.
⚄ The hereditary, myopathy diseases are clearly distinct from inclusion body myositis: these are different diseases.
⚄ Lilleker et al. (2024) recommend in the future researchers
and doctors only use “IBM” to specify inclusion body myositis,
and drop the terms “sporadic” and “familial.”
≻ Further, “hIBM” should be dropped and the names of
each specific disease should be used e.g. Myofibrillary myopathy; GNE
myopathy; UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase;
MSP-1; MSP-2; etc..
⚃ 2.2.4.2 Myositis-specific auto-antibodies
(MSAs):
The classification of muscle diseases has been evolving over time as more
and more research discoveries are made.
≻ Different diseases are being recognized and categories adjusted.
≻ Muscle diseases have been classified mainly based on their
clinical presentation.
≻ Today, the recognition of antibodies associated with different
muscle diseases has impacted classification.
≻ “A major advancement in the field of myositis was the
discovery of auto-antibodies that are
specific for myositis,
called
myositis-specific auto-antibodies (MSAs);
(present in up to 60% of patients with IIM), which are helpful in
establishing a diagnosis of IIM (Lundberg et al., 2021).
≻ Other patients may have auto-antibodies that are also present in
other autoimmune disorders, such as systemic lupus erythematosus (SLE),
systemic sclerosis, or Sjögren syndrome; these auto-antibodies are named
myositis-associated auto-antibodies (MAAs).
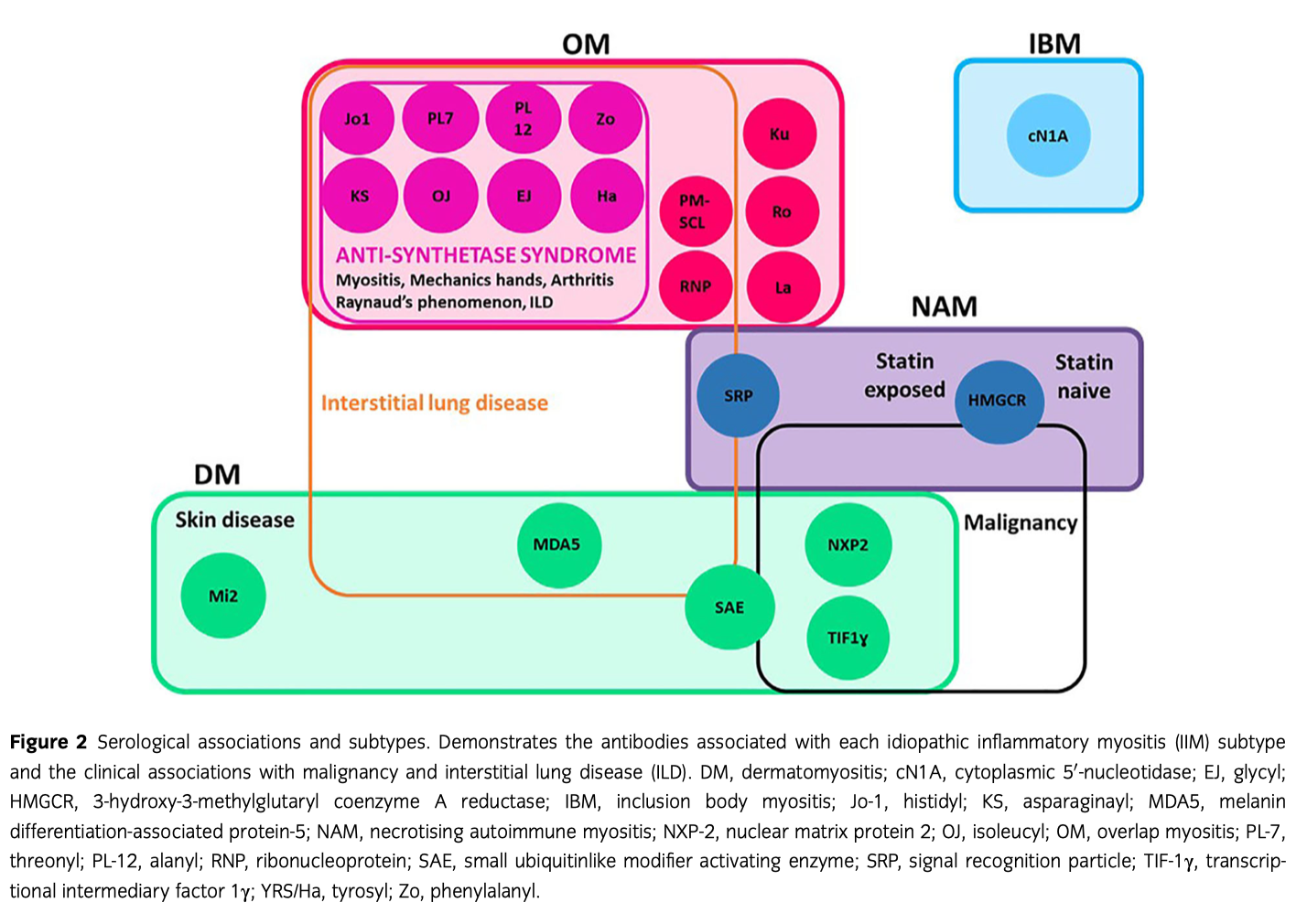
Figure 2.2.4.2.1a
Figure from Ashton, C., Paramalingam, S., Stevenson, B., Brusch, A., & Needham, M. (2021). Idiopathic inflammatory myopathies: A review. Internal Medicine Journal, 51 (6), 845-852. Source.

Figure 2.2.4.2.1b
Figure 2.2.4.2.1b is based on: Mariampillai, K., Granger, B., Amelin, D., Guiguet, M., Hachulla, E., Maurier, F., … Benveniste, O. (2018). Development of a new classification system for idiopathic inflammatory myopathies based on clinical manifestations and myositis-specific autoantibodies. JAMA Neurology, 75 (12), 1528. JAMA Neurol. 2018;75(12):1528-1537. doi:10.1001/jamaneurol.2018.2598
Notes on Figure 2.2.4.2.1b
^ trigger: statin use
* trigger: malignancy
**
CN1A [Anti-NT5c1A] – [anti-cN1A]:
Found in approximately 50 to 60% of IBM patients. Also found in about 25
percent of patients with juvenile myositis and in about 12 percent of
healthy children (Yeker et al., 2018).
≻ According to
Greenberg (2019),
"Anti-cN1A antibodies
are
highly specific to IBM
and are seen in 90-95% of patients with IBM compared with 5-10% of
patients with polymyositis, dermatomyositis or non-immune neuromuscular
diseases."
≻ However, these antibodies have
only moderate diagnostic sensitivity,
ranging from 37% to 76%.
≻ Varying sensitivities may be related to the different methods of
testing that have been used.
≻ In other words, these antibodies are very specific to IBM –
if you have them, you probably have IBM but, they are not that sensitive
to IBM – only about 50 to 60% of IBM patients will have these
antibodies – if you
do not
have them, you may still have IBM.
⚄ Anti-NT5C1A autoantibodies were detected in 71 (61%) of 117 patients with IBM, 2 (5%) of 42 patients with PM, 2 (5%) of 42 healthy volunteers, 24 (15%) of 159 patients with DM, 10 (23%) of 44 patients with Sjögren's syndrome, and 13 (14%) of 96 patients with systemic lupus erythematosus" (Lloyd et al., 2016).
⚄
Anti-cN-1A autoantibodies
are demonstrated in one third of the patients with sIBM and in less than
5% with other IIMs or neuromuscular diseases.
≻ A recent study demonstrated that positive
anti-cN-1A
sIBM patients are included in a more severe sIBM subtype and represent a
homogeneous group as exhibiting higher mortality risk, less proximal upper
limb weakness (not typical of sIBMs) and a cytochrome oxidase deficiency
in muscular fibers, when compared to negative patients" (Palterer et
al., 2018).
⚄ For information on myositis-associated autoantibodies see: https://understandingmyositis.org/myositis-antibody-testing/