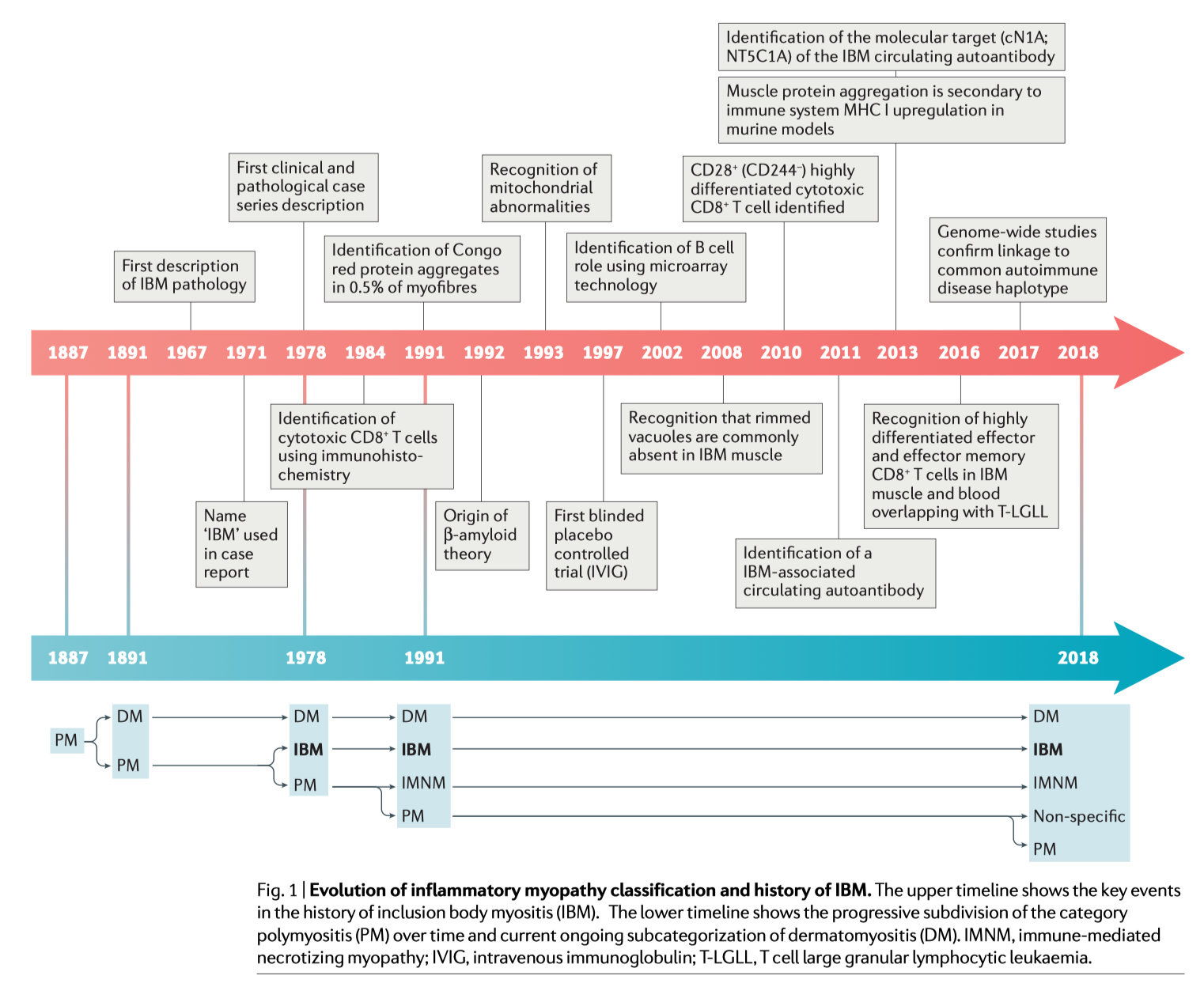
⚃ From: Greenberg, S. A. (2019). Inclusion body myositis: Clinical features and pathogenesis. Nature Reviews Rheumatology 2019, 1. https://www.nature.com/articles/s41584-019-0186-x
⚄ Early descriptions of inclusion body myositis are fairly disjointed and some did not reflect the disease we know today as inclusion body myositis.
⚄
1965: First case report made by Adams, Kakulas and Samaha who
described inclusions in the muscles of a 20-year-old male (Kagen, 2009).
≻ Adams, Kakulas and Samaha provide a description of nuclear
and cytoplasmic inclusions in the muscle of a
20-year-old male student
with a syndrome of progressive weakness of the extremities and trunk.
Finger flexor strength was spared. Serum creatine kinase activity was
mildly elevated. Biopsy of muscle demonstrated the presence of histiocytes
and lymphocytes distributed perivascularly, interstitially, and around
necrotic fibers. Eosinophilic inclusions were present in the cytoplasm of
myofibers and in sarcolemmal nuclei.
⚄ 1967: Chou described inclusions in the muscle cells of a man with "chronic polymyositis" who displayed distinctive features on biopsy, progressive weakness, dysphagia, and muscle atrophy. Electromyograms suggested myositis, although serum enzyme levels were not significantly elevated. A mononuclear cell infiltrate was found in muscle on biopsy.
⚄ 1970: A case reported by Carpenter and Karpati of a 39-year-old woman, with a progressive myopathy for 10 years. The onset of her weakness, which affected both proximal and distal muscles, was insidious. Her forearm and hand muscles, however, were strong. Serum creatine kinase values were normal. Cytoplasmic and nuclear filamentous inclusions were found in muscle on biopsy. In addition, this report called attention to the presence of vacuoles within the cytoplasm of myofibers.
⚄ 1971: Yunis & Samaha first used the term “inclusion body myositis.” They described biopsy findings in the muscle of a 26-year-old woman with weakness of both upper and lower extremities that developed over the course of several years. Although there was severe wasting in multiple areas, the strength of the quadriceps in the lower extremities and of the muscles of the hands was preserved. Serum creatine kinase levels were normal. Biopsy of muscle revealed the "typical picture of chronic polymyositis." Again, nuclear and cytoplasmic inclusions were found with a "focal and mild diffuse round cell infiltration."
⚄ 1975: Mitochondrial abnormalities in IBM muscle identified by Carpenter and Karpati.
⚄ These early reports led to the recognition that inclusion body myositis might be a distinct disease (Kagen, 2009).
⚄ 1977: Early reference to inclusion body myositis as a viral disease (Ketelsen).
⚄ 1978: Carpenter and Karpati describe the abnormal filaments in muscle cells necessary for definite diagnosis. First mention that IBM does not improve with corticosteroid treatment.
⚄ 1981: Nonaka and his team described a form of muscular disorder essentially the same as autosomal recessive inclusion body myopathy type 2 (IBM2).
⚄ 1984: Argov and Yarom describe hereditary Inclusion Body Myopathy (Autosomal Recessive; IBM2) in Jews of Persian origin, characterized by lack of inflammation and quadriceps sparing. They use the term myopathy to distinguish it from the spontaneous myositis form characterized by inflammation.
⚄ 1988: Karpati describes how MHC class I molecules are expressed on myofibers in s-IBM.
⚄ 1988: Arahata and Engel identify evidence of cell-mediated cytotoxicity showing that autoinvasive CD8+ T cells surround MHC class I-immunoreactive myofibers and express perforin and other markers of activation.
⚄ 1989: Lotz reviewed 40 cases and concluded that IBM should be considered a distinct disease. Data were consistent with previous observations that corticosteroid therapy (usually prednisone) does not work in IBM.
⚄ 1990: Baumbach reports the first familial cases of inclusion body myositis.
⚄ 1991: Mendell describes using Congo red stain to detect the presence of amyloid in the inclusions found in IBM muscle cells.
⚄ 1992: Askanas, Engel and Alvarez describe beta-amyloid protein in muscle biopsies of patients with inclusion-body myositis.
⚄ 1992: Askanas & W. K. Engel outline the beta-amyloid theory of IBM.
⚄ 1993: Askanas and Engel note the similarities between the pathology of sIBM muscle and brains affected by Alzheimer's disease: the accumulation of ubiquitin, beta-amyloid protein and its precursor protein, alpha 1-antichymotrypsin, and hyperphosphorylated tau.
⚄ 1993: Askanas and Engel: “In 1993 we introduced the term hereditary inclusion-body myopathies to designate hereditary muscle diseases with muscle pathologic features strikingly resembling those of sporadic IBM, except for a lack of lymphocytic inflammation” (Askanas & Engel, 1998).
⚄ 1993: Oldfors describes mitochondrial abnormalities in IBM.
⚄ 1994: Garlepp reports sIBM is associated with a group of genes linked to the immune system called HLA-DR3 and with the extended 8.1AH. This group of genes is associated with some of the most common autoimmune diseases and is relatively abundant people in the western hemisphere.
⚄ 1995: Griggs describes diagnostic criteria for IBM, specifically emphasizing the presence of characteristic rimmed vacuoles with evidence of the beta-amyloid fragment of the beta-amyloid precursor protein and ubiquitin protein.
⚄ 1996: Mitrani-Rosenbaum S, et al establish genomic linkage of hereditary inclusion body myopathy (hIBM-2) to chromosome 9p1-q1among Iranian Jews.
⚄ 1996: Mirabella demonstrates antibodies (SMI-31) react with paired-helical filaments (PHFs) that characterize sIBM and the hereditary inclusion-body myopathies. SMI-31 can be used for identifying and distinguishing s-IBM and the h-IBMs.
⚄ 1998: The first book on IBM is published, Askanas, Serratrice, and Engel, editors.
⚄ 1998: Darin describes autosomal dominant inclusion body myopathy 3, (IBM3) linked to mutations in a gene producing myosin heavy chain II proteins on chromosome 17.
⚄ 1999: Eisenberg localizes the gene for Inclusion Body Myopathy 2 (Autosomal Recessive; IBM2) in Middle Eastern Jews.
⚄ 2002: Sugarman. The first animal model of inclusion body myositis is developed. The report also notes that the accumulation of amyloid-beta peptide, which is derived from the larger amyloid-beta precursor protein (betaAPP), seems to be an early pathological event in Alzheimer's disease and in sIBM.
⚄ 2002: Tawil improves diagnostic criteria.
⚄ 2004: Price shows another group of genes linked to the immune system, the 35.2AH appear to be associated with sIBM in Caucasians.
⚄ 2004: Fratta reports that the majority of muscle fibers in sporadic inclusion body myositis contain strong immunoreactivity to mutant ubiquitin (UBB+1). Fratta suggests that the UBB+1-inhibited proteasome cannot properly degrade toxic proteins, resulting in their accumulation and aggregation.
⚄ 2005: A major conference on s-IBM. 22 articles were published in Neurology 66 (2) Supplement 1 January 24, 2006.
⚄ 2006: Askanas & W. K. Engel, describe beta amyloid protein in IBM.
⚄ 2006: Dalakas found in addition to the vacuoles and inclusions, muscle tissue in inclusion body myositis shows an inflammatory invasion of intact muscle cells by macrophages and cytotoxic CD8 + T cells.
⚄ 2007: Askanas & W. K. Engel point out that muscle cell degeneration is characterized by progressive cell death, of the development of vacuoles, and the accumulation of clumps made up of different proteins—amyloid inclusion bodies. The amyloid inclusions are of two types, one of which contains beta amyloid protein, and the other, tau protein.
⚄ 2008: Chahin stated that rimmed vacuoles are absent in 20% of patients with typical clinical features of IBM.
⚄ 2008: Jackson introduces the IBM functional rating scale https://doi.org/10.1002/mus.20958.
⚄ 2008: TDP-43 is identified by Weihl and colleagues in non-nuclear sarcoplasm in sIBM and hereditary inclusion body myopathy (hIBM) due to VCP mutations.
⚄ 2009: The "beta amyloid hypothesis of IBM" is taken to task by Greenberg. Greenberg (2009 Curr Neurol Neurosci Rep.) addressed limitations in the beta-amyloid-mediated theory of IBM myofiber injury, flawed rationales of animal models of this disease, and recent reports regarding treatment. Greenberg (2009 Br Med J) demonstrated that distortions in the citation of articles concerning beta-amyloid as created a false impression of the possible role of beta-amyloid in IBM.
⚄ 2009: Gene therapy using follistatin to inhibit myostatin holds promise for the treatment of muscle disease. (Rodino-Klapac, 2009).
⚄ 2009: The first report of specific muscle protein being reduced in IBM muscle: substantial depletion in fast-twitch sarcomeric and glycolytic enzyme proteins in sIBM samples. (Parker, 2009).
⚄ 2009: Following up on earlier leads, the Greenberg group report finding that the protein TDP-43 is a prominent and highly sensitive and specific feature of IBM. This protein is normally found within the nucleus but in IBM is found in the cytoplasm of the cell. (Salajegheh, 2009).
⚄ 2010: Pandya identified CD28 + (CD244 - ) a highly differentiated cytotoxic CD8 + T cell in IBM.
⚄ 2011: Salajegheh (of the Greenberg group) discovered that a circulating autoantibody against a 43-kDa muscle autoantigen is present in IBM.
⚄ 2013: Rose presents The European Neuromuscular Centre (ENMC) diagnostic criteria for IBM.
⚄ August 20, 2013: Novartis receives FDA breakthrough therapy designation for BYM338 (bimagrumab) for sporadic inclusion body myositis (sIBM)
⚄ 2013: The target autoantigen of the IBM [auto]antibody discovered in 2011 was identified simultaneously by two research groups and the findings published in tandem: Larman (of the Greenberg group), Pluk (of the Badrising group) Cytosolic 5' -nucleotidase 1A (cN1A; NT5C1A).
⚄ 2016: Greenberg recognizes highly differentiated effector and effector memory CD8 + T cells in IBM muscle and blood overlapping with T cell large granular lymphocytic leukaemia.
⚄ 2017: Lilleker described differences in clinical and histopathological features between anticytosolic 5'-nucleotidase 1A antibody positive and negative IBM patients.
⚄ 2017: Rothwell reported alleles in the HLA-DRB1 (an autoimmune haplotype) were found to be independently associated with IBM.
⚄ 2018: Britson found variants within the HLA locus are genetic risk factors for developing IBM.
⚄ 2018: Britton noted 65% to 80% of IBM patients have dysphagia and associated issues with excessive thick mucus.
⚄ 2018: Felice reported the anti-cN1A antibody test has a low predictive value for IBM parameters. In our cohort, 20 of 40 (50%) of patients tested positive for anti-cN1A, and, of these, antibodies were strong positive in 12 (60%), moderate positive in 5 (25%), and weak positive in 3 (15%). … Based on all clinical studies published to date including the present, the anti-cN1A antibody test shows low predictive value in regards to disease severity and associated clinicopathological findings.
⚄ 2019: Greenberg (2019) pointed out that, despite the finding that less than one percent of muscle cells contain abnormal proteins, for some reason, the degenerative aspects of IBM have dominated past research. Although immune system involvement was seen early on, it has not received as much research attention. Today, mounting evidence that IBM is an autoimmune T cell-mediated disease provides hope for the development of new, immune based therapies.
⚄ 2019: Greenberg et al., (2019) described the specific cytotoxic CD8 + T cells responsible for muscle cell destruction in IBM and a marker of these cells (KLRG1).
⚄ 2019: Keshishian et al., found that IBM patients were more likely to have multiple comorbidities, including cardiovascular risk factors and events, muscle and joint pain, and pulmonary complications compared to those without IBM. https://doi.org/10.1080/03007995.2018.1486294.
⚄ 2019: Ramdharry et al., presents the latest version of the clinician rated inclusion body myositis functional rating scale.
⚄ 2020: Cantó-Santos et al., discussed the mechanisms underlying energy production, oxidative stress generation, cell signaling, autophagy, and inflammation triggered or conditioned by the mitochondria. mtDNA deletions are reported in 67% of sIBM patients.
⚄ 2020: Paul et al., reported that anti-cN1A antibodies do not correlate with specific clinical, EMG or pathological findings in sporadic inclusion body myositis.
⚄ 2021: Lelièvre et al., described functional impacts of diaphragmatic involvement in patients with inclusion body myositis.
⚄ 2021: Based upon the rationale presented in Greenberg et al., (2019), Abcuro Inc. has developed a monoclonal antibody designed to target KLRG1+, a marker of highly differentiated cytotoxic T cells. This antibody named ABC008 is currently in clinical trials, being administered subcutaneously to IBM patients. Preliminary results (November 2021) demonstrate proof of mechanism and more trials are planned. Clinical trial information. More information.
⚄ 2021: Phase III study: Sirolimus (also known as rapamycin) is an immunosuppressive agent, currently used together with other medicines to prevent the body from rejecting a transplanted kidney. The hypothesis is that Sirolimus, will block the activity of T effector cells but preserving T regulatory cells, as well as by inducing autophagy (protein degradation), will be effective in IBM to slow or stabilize disease progression, helping to maintain patient function and independence. This phase III trial will confirm pilot data showing statistically significant clinical outcomes. Principal Investigator: Mazen Dimachkie, University of Kansas Medical Center. Clinical trial information.
⚄ 2021: Lundberg et al., presents a review of inclusion body myositis. https://www.nature.com/articles/s41572-021-00321-x
⚄ 2021: Jørgensen et al., emphasized that blood flow restricted exercise can play a big role in helping IBM patients. https://doi.org/10.1111/sms.14079.
⚄ 2021: Naddaf et al., showed that on average, IBM patients die three years younger, mostly due to the presence of dysphagia or respiratory failure/pneumonia. As well, that study showed IBM patients treated with corticosteroids had lower survival rates. https://doi.org/10.1093/rheumatology/keab716
⚄ 2021: Shelly et al., emphasized that other autoimmune disorders are more common in IBM patients (rheumatoid arthritis, Sjogren's syndrome) but that the risk of cancer did not increase in IBM. Life expectancy was lower in female patients, patients with dysphagia, and patients that developed IBM after the age of 67. https://doi.org/10.1212/WNL.0000000000012004.
⚄ 2022: Goyal showed that KLRG1+ CD8+ T cells are highly differentiated cells that are over-represented in the blood of patients with IBM and that a monoclonal antibody should kill KLRG1 without compromising the ability of regulatory T cells to suppress autoimmunity. https://pubmed.ncbi.nlm.nih.gov/35131904/
⚄ 2022: Britson et al., took muscle cells from IBM patients and put them inside the legs of humanized mice. These cells died, but the mouse muscle then showed regeneration of new muscle cells that continued to show the usual features of IBM disease, including invading human KLRG1+ T cells, rimmed vacuoles, and the abnormal accumulation of proteins, including TDP-43. The mice were then treated with OKT3 to kill the invading human T cells. Although this treatment reduced the number of invading human KLRG1+ T cells by 96%, the newly generated human-like muscle cells still showed the distinctive IBM features. https://doi.org/10.1126/scitranslmed.abi9196.
⚄ 2023: Michelle et al., showed Black and female patients represent clinically distinct subgroups within IBM with unique disease trajectories and, potentially, different responses to therapeutic interventions.
⚄ 2023: Cantó-Santos et al., Findings confirm the presence of molecular disturbances in peripheral tissues from IBM patients and prompt patients’ derived fibroblasts as a promising disease model, which may eventually be exported to other neuromuscular disorders. We additionally identify new molecular players in IBM associated with disease progression, setting the path to deepen in disease aetiology, in the identification of novel biomarkers or in the standardization of biomimetic platforms to assay new therapeutic strategies for preclinical studies.
⚄ 2023: Lee et al., Clinically significant differences were not found between anti-NT5c1A antibody-seropositive and seronegative IBM groups with respect to gender, age at symptom onset, age at diagnosis, disease duration, serum CK values, presence of other autoantibodies, dysphagia, and the pattern of muscle impairment.
⚄
2023: Cantó-Santos, J., Valls-Roca, et al., Metabolic dysregulation
in IBM is present outside the target tissue (muscle), as seen in the
altered organic acids in fibroblasts and urine;
The detection of L-pyroglutamic and orotic acids in urine displayed an
outstanding biomarker signature, with 100% sensitivity and specificity
⚄ 2023: Vogt et al., Despite HIV-IBM and sIBM sharing important clinical, histopathological, and transcriptomic signatures, the presence of KLRG1 cells discriminated sIBM from HIV-IBM.
⚄
2023: Lindgren et al., The mean age at symptom onset was 38 years
and mean age at diagnosis was 45 years in patients with early-onset IBM,
while mean age at symptom onset was 64 years and mean age at diagnosis was
70 years in the corresponding IBM cohort.
Early-onset IBM is a severe inflammatory myopathy, causing progressive
muscle weakness, high mitochondrial DNA mutation load in muscle fibers and
a reduced cumulative survival in young and middle-aged individuals.
⚄ 2023: Nelke et al., Tissue-resident FAPs, not myofibers, are the main cell type assuming a senescent phenotype in IBM.
⚄ 2023: Tanboon et al., PM with mitochondrial pathology (PM-Mito), which contains features resembling IBM except for rimmed vacuoles (i.e., endomysial inflammation, mitochondrial pathology, highly differentiated cytotoxic T cells, and type 2 interferon (IFN2, IFN-Y) pathway upregulation) has been re-recognized and proposed as early IBM (eIBM) in IBM-spectrum disease.
⚃ References:
⚄ If the reference does not appear here you will find it in the research section under appropriate year ( LINK )
⚄ Adams RD, Kakulas BA, Samaha FA. A myopathy with cellular inclusions. Trans Am Neurol Assoc 1965;90:213-6.
⚄ Arahata K, & Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies. V: Identification and quantization of T8+ cytotoxic and T8+ suppressor cells. Ann Neurol. 1988 May;23(5):493-9.
⚄ Argov Z. & Yarom, R., "Rimmed vacuole myopathy" sparing the quadriceps. A unique disorder in Iranian Jews. J Neurol Sci. 1984 Apr;64(1):33-43.
⚄ Argov Z, Tiram E, Eisenberg I, Sadeh M, Seidman CE, Seidman JG, Karpati G, Mitrani-Rosenbaum S. Various types of hereditary inclusion body myopathies map to chromosome 9p1-q1.Ann Neurol. 1997 Apr;41(4):548-51. Comment in: Ann Neurol. 1997 Apr;41(4):421-2. Hereditary inclusion body myopathies are a clinically heterogeneous group of disorders characterized by adult-onset, slowly progressive muscle weakness and typical histopathology: rimmed vacuoles and filamentous inclusions. The disorders are usually inherited as an autosomal recessive trait. The gene responsible for the disease found in Iranian Jews, who present with quadriceps-sparing myopathy, maps to chromosome 9p1-q1. We address the question of whether hereditary inclusion myopathies are genetically as well as clinically heterogeneous disorders. We mapped the disease gene segregating in two families of Afghani-Jewish and one family of Iraqi-Jewish descent to the chromosome 9 locus. Similarly, the disease gene segregating in a non-Jewish family from India mapped to the same locus. By contrast, the disease gene segregating in a French-Canadian family in which affected individuals had central nervous system involvement as well as hereditary inclusion body myopathy, did not map to this locus. We conclude that many but not all forms of autosomal recessive hereditary inclusion body myopathy are caused by a gene defect that maps to chromosome 9p1-q1.
⚄ Askanas, V., & Engel, W. K. (1998). Sporadic Inclusion-Body Myositis and Hereditary Inclusion-Body Myopathies. Archives of Neurology, 55 (7), 915. doi:10.1001/archneur.55.7.915
⚄ Askanas V, Engel WK. New advances in inclusion-body myositis. Curr Opin Rheumatol. 1993 Nov;5(6):732-41.
⚄ Askanas, V., Serratrice, G., Engel, W. K. (Eds.). (1998). Inclusion-Body Myositis and Myopathies. United Kingdom: Cambridge University Press.
⚄ Askanas, V. & Engel, W. K., Inclusion-body myositis: a myodegenerative conformational disorder associated with A-beta, protein misfolding, and proteasome inhibition. Neurology 66 (suppl. 1): S39-S48, 2006.
⚄ Askanas V, Engel WK, & Alvarez RB. Light and electron microscopic localization of beta-amyloid protein in muscle biopsies of patients with inclusion-body myositis. Am J Pathol. 1992 Jul;141(1):31-6.
⚄ Carpenter S, Karpati G, Wolfe L. Virus-like filaments and phospholipid accumulation in skeletal muscle. Neurology 1970; 20: 889-903.
⚄ Carpenter S, Karpati G, & Eisen A. A morphologic study in polymyositis: clues to pathogenesis of different types. In: Bradley W, ed. Recent Advances in Myology. Amsterdam: Excerpta Medica, 1975:374-379.
⚄ Carpenter S, Karpati G, Heller I, & Eisen A. Inclusion body myositis: a distinct variety of idiopathic inflammatory myopathy. Neurology. 1978 Jan;28(1):8-17.
⚄ Chou SM. Myxovirus-like structures in a case human chronic polymyositis. Science 1967;158:1453-5.
⚄ Chou SM. Myxovirus-like structures and accompanying nuclear changes in chronic polymyositis. Arch Pathol 1968; 86:649-6.
⚄ Dalakas, M. C., Sporadic Inclusion Body Myositis-Diagnosis, Pathogenesis and Therapeutic Strategies. Nat Clin Pract Neurol. 2006;2(8):437-447.
⚄ Darin, N.; Kyllerman, M.; Wahlstrom, J.; Martinsson, T.; Oldfors, A. Autosomal dominant myopathy with congenital joint contractures, ophthalmoplegia, and rimmed vacuoles. Ann. Neurol. 44: 242-248, 1998.
⚄ Eisenberg I, Thiel C, Levi T, Tiram E, Argov Z, Sadeh M, Jackson CL, Thierfelder L, Mitrani-Rosenbaum S. Fine-structure mapping of the hereditary inclusion body myopathy locus. Genomics. 1999 Jan 1;55(1):43-8.
⚄ Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, Barash M, Shemesh M, Sadeh M, Grabov-Nardini G, Shmilevich I, Friedmann A, Karpati G, Bradley WG, Baumbach L, Lancet D, Asher EB, Beckmann JS, Argov Z, Mitrani-Rosenbaum S. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001 Sep;29(1):83-7.
⚄ Fratta, P.; Engel, W. K.; Van Leeuwen, F. W.; Hol, E. M.; Vattemi, G.; Askanas, V.: Mutant ubiquitin UBB+1 is accumulated in sporadic inclusion-body myositis muscle fibers. Neurology 63: 1114-1117, 2004.
⚄ Garlepp MJ, Laing B, Zilko PJ, Ollier W, Mastaglia FL. HLA associations with inclusion body myositis. Clin Exp Immunol 1994; 98:40-5.
⚄ Greenberg SA. Inclusion body myositis: review of recent literature. Curr Neurol Neurosci Rep. 2009 Jan;9(1):83-9.
⚄ Greenberg SA. How citation distortions create unfounded authority: analysis of a citation network. Br Med J 2009; 339:b2680.
⚄ Griggs RC, Askanas V, DiMauro S, et al. Inclusion body myositis and myopathies. Ann Neurol 1995; 38: 705-13.
⚄ Ikeuchi T, Asaka T, Saito M, Tanaka H, Higuchi S, Tanaka K, Saida K, Uyama E, Mizusawa H, Fukuhara N, Nonaka I, Takamori M, Tsuji S. Gene locus for autosomal recessive distal myopathy with rimmed vacuoles maps to chromosome 9. Ann Neurol. 1997 Apr;41(4):432-7.
⚄ Karpati G, Pouliot Y, Carpenter S: Expression of immunoreactive major histocompatibility complex products in human skeletal muscles. Ann Neurol 1988 Jan; 23(1): 64-72
⚄ Ketelsen UP, Beckmann R, Zimmermann H, Sauer M. Inclusion body myositis. A "slow-virus" infection of skeletal musculature? Klin Wochenschr. 1977 Nov 1;55(21):1063-6.
⚄ Kitazawa M, Trinh DN, Laferla FM (2008). Inflammation induces tau pathology in inclusion body myositis model via glycogen synthase kinase 3 beta. Ann. Neurol. doi:10.1002/ana.21325. PubMed Link
⚄ Massa R, Weller B, Karpati G, Shoubridge E, Carpenter S. Familial inclusion body myositis among Kurdish-Iranian Jews. Arch Neurol. 1991 May;48(5):519-22. Neuromuscular Research Group, Montreal (Quebec), Neurological Institute, Canada. We report two cases of adult-onset, slowly progressive limb-girdle muscle weakness with a remarkable sparing of quadriceps muscles that developed in patients from different families of Iranian-Kurdish-Jewish origin. Each patient had a similarly affected sibling. The findings by means of muscle biopsies showed abnormalities typical of inclusion body myositis, including abundant lined vacuoles and characteristic cytoplasmic inclusions of 15- to 18-nm filaments. Remarkably, many vacuolated muscle fibers showed immunoreactivity to neural cell adhesion molecule, a fetal muscle antigen. The common origin of these patients from an isolated ethnic group with frequent consanguinity and the familial incidence is indicative of a genetic causation or predisposition, probably with an autosomal recessive inheritance. This familial myopathy is one of several clinical syndromes that share the typical pathological findings of inclusion body myositis. The pathogenic relationship between these different familial forms and the more common sporadic form of inclusion body myositis is not known.
⚄ Mendell JR, Sahenk Z, Gales T, Paul L. Amyloid filaments in inclusion body myositis. Novel findings provide insight into nature of filaments. Arch Neurol. 1991 Dec;48(12):1229-34.
⚄ Mirabella M, Alvarez RB, Bilak M, Engel WK, Askanas V. Difference in expression of phosphorylated tau epitopes between sporadic inclusion-body myositis and hereditary inclusion-body myopathies. J Neuropathol Exp Neurol. 1996 Jul;55(7):774-86.
⚄ Mitrani-Rosenbaum S, Argov Z, Blumenfeld A, Seidman CE, Seidman JG. Hereditary inclusion body myopathy maps to chromosome 9p1-q1. Hum Mol Genet 1996;5:159 -163.
⚄ Morosetti, Roberta, et al, MyoD expression restores defective myogenic differentiation of human mesoangioblasts from inclusion-body myositis muscle PNAS, November 7, 2006, vol. 103, no. 45, 16995-17000.
⚄ Needham M. and Mastaglia FL, (2007) Inclusion body myositis: current pathogenetic concepts and diagnostic and therapeutic approaches. Lancet Neurol 2007; 6: 620-31.
⚄ Nishino I, Noguchi S, Murayama K, Driss A, Sugie K, Oya Y, Nagata T, Chida K, Takahashi T, Takusa Y, Ohi T, Nishimiya J, Sunohara N, Ciafaloni E, Kawai M, Aoki M, Nonaka I. Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology. 2002 Dec 10;59(11):1689-93. Comment in: Neurology. 2002 Dec 10;59(11):1674-6. Neurology. 2003 Jul 8;61(1):145; author reply 145. BACKGROUND: Distal myopathy with rimmed vacuoles (DMRV) is an autosomal-recessive disorder with preferential involvement of the tibialis anterior muscle that starts in young adulthood and spares quadriceps muscles. The disease locus has been mapped to chromosome 9p1-q1, the same region as the hereditary inclusion body myopathy (HIBM) locus. HIBM was originally described as rimmed vacuole myopathy sparing the quadriceps; therefore, the two diseases have been suspected to be allelic. Recently, HIBM was shown to be associated with the mutations in the gene encoding the bifunctional enzyme, UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE). OBJECTIVE: To determine whether DMRV and HIBM are allelic. METHODS: The GNE gene was sequenced in 34 patients with DMRV. The epimerase activity in lymphocytes from eight DMRV patients was also measured. RESULTS: The authors identified 27 unrelated DMRV patients with homozygous or compound-heterozygous mutations in the GNE gene. DMRV patients had markedly decreased epimerase activity. CONCLUSIONS: DMRV is allelic to HIBM. Various mutations are associated with DMRV in Japan. The loss-of-function mutations in the GNE gene appear to cause DMRV/HIBM.
⚄ Nonaka, I.; Sunohara, N.; Ishiura, S.; Satoyoshi, E. Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J. Neurol. Sci. 51: 141-155, 1981. Price P, Santoso L, Mastaglia F, et al. Two major histocompatibility complex haplotypes influence susceptibility to inclusion body myositis: critical evaluation of an association with HLA-DR3. Tissue Antigens 2004;64:575-80.
⚄ Parker KC, Kong SW, Walsh RJ; Bch, Salajegheh M, Moghadaszadeh B, Amato AA, Nazareno R, Lin YY, Krastins B, Sarracino DA, Beggs AH, Pinkus JL, Greenberg SA. Fast-twitch sarcomeric and glycolytic enzyme protein loss in inclusion body myositis.Muscle Nerve. 2009 Mar 16. [Epub ahead of print]
⚄ Rodino-Klapac LR, Haidet AM, Kota J, Handy C, Kaspar BK, Mendell JR. Inhibition of myostatin with emphasis on follistatin as a therapy for muscle disease.Muscle Nerve. 2009 Feb 10;39(3):283-296. [Epub ahead of print]
⚄ Sadeh M, Gadoth N, Hadar H, Ben-David E. Vacuolar myopathy sparing the quadriceps. Brain. 1993 Feb;116 ( Pt 1):217-32.
⚄ Salajegheh, M, Pinkus, JL, Taylor, JP, Amato, AA, Nazareno, R, Baloh, RH, Greenberg, SA. Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve. 2009; 40(1):19 -31.
⚄ Sugarman MC, Yamasaki TR, Oddo S, Echegoyen JC, Murphy MP, Golde TE, Jannatipour M, Leissring MA, LaFerla FM. Inclusion body myositis-like phenotype induced by transgenic overexpression of beta APP in skeletal muscle. Proc Natl Acad Sci U S A. 2002 Apr 30;99(9):6334-9.
⚄ Tawil R, Griggs RC. Inclusion body myositis. Curr Opin Rheumatol 2002; 14: 653-57.
⚄ Tomimitsu H, Ishikawa K, Shimizu J, Ohkoshi N, Kanazawa I, Mizusawa H. Distal myopathy with rimmed vacuoles: novel mutations in the GNE gene. Neurology. 2002 Aug 13;59(3):451-4. Comment in: Neurology. 2002 Dec 10;59(11):1674-6. The authors present three novel missense mutations in the UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) gene, the causative gene for hereditary inclusion body myopathy, in Japanese patients with distal myopathy with rimmed vacuoles. Seven out of nine patients had homozygous V572L mutation, one was a compound heterozygote with C303V and V572L mutations, and the remaining patient bore homozygous A631V mutation.
⚄ Weihl CC, Temiz P, Miller SE, et al. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry 2008; 79:1186-1189. This study found sarcoplasmic TDP-43, a protein normally seen only in nuclei, in biopsy samples from patients with inclusion body myositis and VCP mutation inclusion body myopathies.
⚄ Yunis EJ, Samaha FJ. Inclusion body myositis. Lab Invest 1971; 25:240-8.